Purpose grown Radiata Pine forest – New Zealand

Acetylation. What is it and what is acetylated wood?
23.01.2019
An explanation of the mysterious wood technology behind Accoya® wood
If you search online there are academic journals and infamous online encyclopaedic entries that can answer the question ‘what is acetylation’ in a very scientific way. But rather than spin tales of hydroxyls, hydrogen atoms and other chemistry jargon, in the simplest terms we can muster (in the context of our wood technology) acetylation for us is:
Subjecting a softwood to a vinegar, which turns it into a hardwood by preventing the cells in the wood from being able to absorb water.
So acetylated wood is… Pickled wood?
More or less. Yes.
Ok the chemistry behind ‘turns it into’ is a little complex, and the vinegar is acetic anhydride. Not quite the malt vinegar for your fish and chips. But the principle is there. If you do want to know the chemistry behind acetylation, feel free to get in touch.
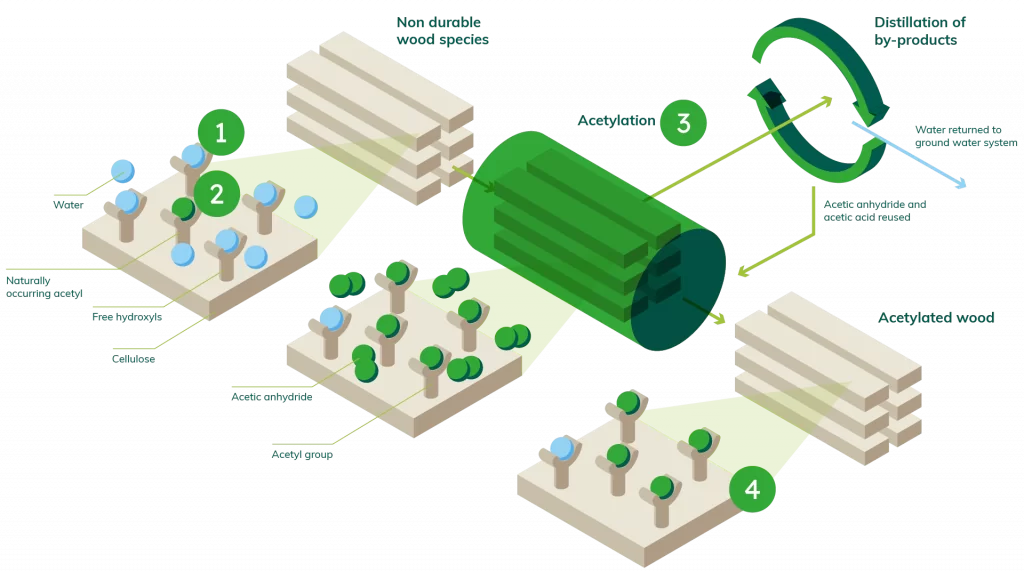
Acetic anhydride? Is that some toxic chemical?
It’s a bit like really strong vinegar in its concentrated form – but it’s actually not present in Accoya wood! It interacts with the wood in our production process to make the wood molecules so resistant to binding with water, and the liquid left over really is just strong vinegar. We even take this by-product out to be used in other industries, or even reprocessed back so we can use it again because we love the circular economy and don’t like waste at all!
So, while you wouldn’t want to put concentrated acetic anhydride on your potato chips, it’s fantastic for making great, long-lasting and safely non-toxic wood products that can last a lifetime.
Why go to the effort of pickling, acetylating your wood?
To understand the benefits of using acetylated timber, we should point out the importance of acetylated wood not absorbing water.
When making anything out of wood, its water that’s one of the main trouble makers.
Keep wood dry and well maintained and it’s one of the greatest materials of all time. But introduce water? Problems begin to seep in. With untreated, badly treated or unmodified wood, water can bring a whole host of problems including rotting, warping, splintering, swelling, shrinking and being food for hungry insects.
So acetylation wood technology removes the issue at source; rather than treating wood with a toxic chemical trying to stop rot or insect attack and not helping with other helping with other water issues. The result is real wood that doesn’t rot. It doesn’t warp, swell or shrink. It doesn’t splinter and it isn’t termite chow. In fact removing all these traditional issues means that acetylated wood presents even more benefits than just ‘not failing’. It is so dimensionally stable (maintains its original shape and dimensions) that it’s perfect for coating and has outstanding durability. This wood, our wood, is branded Accoya®. Accoya® is acetylated wood. In fact Accoya® is the only acetylated wood, and in our opinion the only wood worth considering.
No longer at risk of traditional wood failure gives you confidence of longevity. Meaning any deck, cladding, windows or doors, boat, desk, fence, shed, giant wooden octopus, or whatever else you want to make, it’s going to last so much longer than any traditional alternative. So Accoya® is an investment in the future.
No water. No movement. No problem!
Sounds great, what’s the catch with this new-fangled technology?
Actually the technology isn’t new at all. Acetylation has been around for at least most of the 20th century. The problem is that it’s relatively easy to do it on a small scale, but very difficult to do large scale. It’s only recently that we’ve been able to acetylate larger volumes of wood consistently. So it has only been commercially viable and available since around 2007.
And there’s absolutely no catch. It’s a wonder material. Reliable, eco-friendly and sustainable.

How is using wood sustainable? Aren’t we cutting down the rainforests to get this wood?
We source all our wood from sustainable wood farms. So all the wood used is purposefully grown. This means we increase biomass; our acetylated wood is a carbon sink, good for the environment.
Cutting down rainforests is an issue with unsustainably sourced wood, and a problem with some traditional hardwoods.
An exotic hardwood tree may take hundreds of years to grow. Vast swathes of rainforest are cut down to find these old trees, to be turned into someone’s deck. Resulting in a huge carbon footprint and the destruction of natural resources.
What we want to do is provide even higher quality wood, but without any of the negative impact. Plus hardwoods aren’t all they’re cracked up to be. Oak, Teak, etc will still warp, rot, splinter, etc. Acetylated wood won’t.
Plus regardless of responsibly, sustainably sourced or not, most wood carries a carbon footprint higher than Accoya®. Our acetylated wood has an exceptionally low carbon footprint compared with alternatives.
So rounding back to the original question of what is acetylation? Technically it’s a chemical process. But we could say that acetylation is an opportunity for peace of mind, to invest in our homes and protect our planet.

Where to Buy
You can buy Accoya and Accoya products from our selection of distributors or manufacturers in your region. Use our map search tool to find your nearest Accoya supplier.
Go back
WHERE TO BUY ACCOYA
for your next projectWhat type of Product are you interested in?
- - Select product type -
-
Go back
FIND A SUPPLIER
FIND A INSTALLER
for your next projectWhat type of are you interested in? (optional)
How will your be used?
- - Select type -
-
Enter your postcode/location:


Close
You are currently on the Accoya site
Would you like to visit the Accoya Site to view all relevant content for your location?